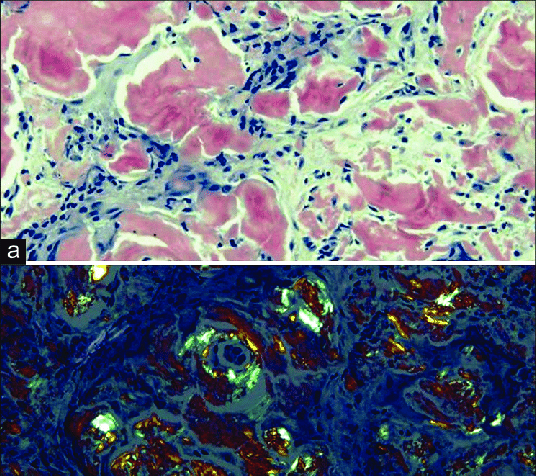
Congo red stain: The gold standard for diagnosing amyloidosis
The gold standard for diagnosis of amyloid is the Congo red stain. All forms of amyloid exhibit affinity for Congo red and give off an apple-green birefringence under polarized light. It is one of the characteristics that distinguish amyloid from other fibrils.
Abstract
Introduction
Amyloidosis is a heterogeneous group of diseases bound by the characteristic deposition of amyloid fibrils in soft tissues.1 Other pathogenic fibrils exist, but only amyloid fibrils are capable of giving off an apple-green birefringence under polarized light after binding to Congo red. To date, > 28 proteins have been identified to be amyloidogenic.2 One of the most recent additions to the amyloid family is ALECT2 (leukocyte cell derived chemotaxin-2).3 As the number of amyloidogenic proteins increases, it outgrew the capacity of the previous nomenclature, which consisted of, for example, primary, secondary, and hereditary.2 The new system uses an abbreviation of the native protein. For example, immunoglobulin light chain is AL, serum amyloid A protein is AA, transthyretin is ATTR, and so on. Although the native proteins come in various sizes and tertiary structures, all amyloid fibrils have the same characteristic cross-β sheet quaternary structure.1 Under x-ray crystallography, amyloid fibrils produce 2 characteristic diffraction signals at 4.7 and 10 angstroms. Fibrillogenesis begins with misfolding of the precursor protein to a lower thermodynamic state. In this state, the intermediate takes on more β-sheet structures that enable aggregation and self-propagation secured by hydrogen bonds. The resultant product then undergoes a process known as steric zipper where the β-sheet regions from different molecules align themselves and interdigitate to form a dry interface.4 The end product is a protofibril, which is more resistant to degradation. Four to 6 protofibrils are weaved together to form an amyloid fibril.
Mechanisms of fibrillogenesis
Amyloidogenesis can occur via a variety of mechanisms.1,2 Several of the more common ones will be discussed to illustrate the diversity of amyloidogenesis. Genetic mutation often with an amino acid substitution can result in an amyloidogenic protein. They represent the hereditary amyloidoses, which include AFib (fibrinogen A α chain), ALys (lysozyme), AApoAI and AApoAII (apolipoprotein AI and AII), and ATTR. Amyloid can also form from wild-type proteins. The best example is senile amyloid of the wild-type transthyretin. Other senile amyloids include APro (prolactin), ACal (calcitonin), AIAPP (amylin), and AANF (atrial natriuretic factor); all are derived of wild-type proteins. Some amyloids form as a result of sustained elevated concentrations of the wild-type protein. This may occur either by increased production as in the case of AA amyloidosis secondary to chronic infectious, inflammatory conditions or Castleman disease, or decreased elimination as in Aβ2m (β2-microglobulin) that occurs in patients with end-stage renal disease. AIns (insulin) is unique in that the injected insulin forms amyloidomas near injection sites via this process. Other ways of increasing amyloidogenicity is proteolytic remodeling. The best example of this is APP (β-amyloid precursor protein) in Alzheimer disease. Some may involve multiple mechanisms (accumulation, mutation, and proteolytic remodeling), such as AL or AH (immunoglobulin heavy chain).5 One unusual mechanism is seen in the familial periodic fever syndromes that include familial Mediterranean fever, TRAPS (TNF receptor–associated periodic syndrome), Muckle-Wells syndrome, and hyper IgD syndrome.6 In familial periodic fever syndromes, the mutations do not code for an amyloidogenic protein but instead cause periodic fevers that lead to the overproduction of serum amyloid A protein resulting in AA amyloidosis. Amyloid can also be transmitted by prions. Prions are the only infectious agents known to contain no nucleic acids. Prions induce disease by acting as a template for the normal prion protein (PrPc) to misfold into the pathogenic PrPsc, which has amyloid features.7 Finally, in some, the mechanism remains undetermined. The best example of this is ALECT2. No mutations, proteolytic remodeling, or elevated levels have been found so far.8,9
Clinical presentation
Amyloidosis presents in a variety of ways and can make diagnosis difficult. This is in part because of the many different types of amyloid, but significant variation can exist within an individual type of amyloid. An example is ATTR. The presence of Val30Met mutation strongly predicts for peripheral neuropathy, autonomic neuropathy, and abnormalities in the cerebrospinal fluid, whereas patients with the Val122Ile mutation are more likely to develop cardiac symptoms, especially in blacks.10,11 It is helpful to separate the presentation into 2 patterns: localized and systemic.1 Localized amyloidoses are confined to 1 site and are generally nonlethal. Skin is a common site of deposition and can manifest as asymptomatic plaques, fissures, or nodules. Examples include lichenoid or macular amyloidosis secondary to AD (keratin) deposition in the skin possibly as a result of trauma.2 Amyloidomas composed of AIns are found near the injection sites of diabetics. AL amyloidosis, which is often systemic, also has a localized variant.12,13 Nodular localized cutaneous (AL) amyloidosis is probably a response to a localized inflammatory reaction often produced by polyclonal plasma cells. Patients commonly present with skin plagues (skin), microscopic or gross hematuria (bladder), and unexplained voice changes (larynx), but amyloid can be deposited in other soft tissues. Progression to systemic AL amyloidosis is rare, but local recurrence is not uncommon.14,15
Systemic amyloidosis implies involvement of visceral organ(s) or multiple tissues. Patients may present with symptoms limited to 1 organ or with multiorgan failure. The most common form of systemic amyloidosis is AL amyloidosis, which outnumbers AA amyloidosis by 20:1 in United States but only 2:1 in Europe because of higher incidence of familial Mediterranean fever.16 Kidney is the most common organ involved in AL, AA, and most forms of hereditary amyloidosis, except ATTR.16–19 Proteinuria is present in 73% of the AL amyloidosis patients, with 30% exhibiting nephrotic syndrome. Renal insufficiency is noted in nearly half of the patients, which is usually associated with proteinuria but can occur without in patients with vascular limited renal amyloidosis.20,21 Renal involvement is nearly universal (97%) in AA amyloidosis.18 Heart is the next most common organ involved in AL amyloidosis with abnormal echocardiographic findings noted in 65% of patients.16 It is the predominate feature in senile TTR amyloidosis.11,19 Presentation varies from asymptomatic to a subtle decrease in exercise capacity to fatigue, dyspnea, and lower extremity edema to angina, syncope, ascites, and anasarca, which are associated with more advanced disease.22 Overt heart failure can be seen in 24%.16 Low voltage on ECG and concentric thickened ventricles on echocardiogram are classic signs of cardiac involvement by amyloidosis. Cardiac involvement, on the other hand, is rare in AA amyloidosis, occurring in only 1% of patients.18 The nervous system is a prominent feature in some forms of amyloidosis and can involve the central nerves, peripheral nerves, or both.16 Peripheral nerve involvement can present with bilateral distal progressive paresthesia or carpel tunnel syndrome. Automatic nervous system involvement is characterized by syncope, erectile dysfunction, gastroparesis, and diarrhea. Some are quite specific as in the case of cystatin c (ACys) amyloidosis, which causes the Icelandic form of hereditary cerebral amyloid angiopathy.2 These patients present with cerebral hemorrhage, stroke, and dementia.2
Symptoms common to most types of amyloid include fatigue and weight loss, which are reported by more than half of the patients.16 Weight loss, however, is unreliable in patients with nephrotic syndrome because many will gain weight as their edema worsens. Gastrointestinal presentation is not common and usually presents with macroglossia, nausea, vomiting, and pseudo-obstruction, which accounts for < 10% of AL amyloidosis cases. Hepatomegaly is present in 24% of AL amyloidosis patients but in only 9% of AA amyloidosis. Bruising is common and often occurs after minor trauma or procedure, especially around the eyes.
Treatment
It is beyond the scope of this article to discuss treatment of amyloidosis in detail. For more comprehensive review of treatment options for AL amyloidosis, interested readers should refer to previous articles.23,24 It is important to recognize that treatments are available for many types of amyloidosis but they are type specific. For localized amyloid, local treatment such as excision or laser removal may be appropriate once systemic disease is ruled out.12 In AL amyloidosis, which is almost always the result of a plasma cell dyscrasia (rarely lymphoproliferative disorders), antimyeloma therapies have been effective.25–28 The treatment of AA amyloidosis is targeted toward the underlying infection or inflammatory condition.18 For patients with a periodic fever syndrome, biologic agents, such as anti-TNF or IL-1 receptor antagonists, have been effective both for symptom control and improvement of the amyloidosis.6,29,30 Organ transplantation has been used in several different types of hereditary amyloidosis.31,32 This is done either to replace damaged organs or to eliminate the source of the mutant protein. The most common example of the latter is liver transplantation in patients with ATTR amyloidosis where the mutant transthyretin is produced primarily in the liver.33 Liver transplantation has also been performed in combination with kidney transplantation to prevent recurrence in the kidney. This has been done successfully in patients with AFib and AApoAI amyloidosis.31,32,34 Although it appears to be effective at preventing recurrence, additional risk of the liver transplantation should be considered.
Because treatment is specific to each type of amyloid, accurate diagnosis is essential. This is especially true when cytotoxic therapy or organ transplantation is being considered. The presence of a monoclonal protein alone is not enough to support the diagnosis of AL amyloidosis or AH amyloidosis. In a single-center study of 350 patients with suspected AL amyloidosis, 34 (10%) were found to have a genetic mutation coding for one of the familial amyloidoses, most commonly AFib and ATTR.35 Eight (24%) of these 34 patients had a monoclonal gammopathy. The number of misdiagnoses could be even greater because some amyloidoses are not the result of genetic mutations. Interestingly, a recent study found that ALECT2 was the most common undiagnosed type of amyloid.9 So far, ALECT2 does not appear to be the result of genetic mutations, but there may be a predilection for homozygosity in the G allele.8 This also applies to senile amyloidosis, which is the result of accumulation of normal transthyretin with no genetic mutations.2
Diagnostic approach
Patients presenting with multiorgan derangement should be screened for amyloidosis. The diagnosis is much more challenging when the patient presents with single-organ involvement as it may mimic more common diseases. A high index of suspicion is often required. Screening should be performed in patients with unexplained heart failure, neuropathy, and nephrotic syndrome. Clues that raise suspicion include low voltage on ECG, severe hypotension without antihypertensive medications, and the presence of neuropathy in combination with other organ involvement.16,22,36 Easy bruisability, especially “raccoon eyes” after procedures or minor trauma, is another hint. Major bleeding can result from factor X and vitamin K-dependent clotting factor deficiency but is much less common.
Several elements of the history should alert the provider toward AA amyloidosis. Chronic inflammatory conditions (eg, rheumatoid arthritis and ankylosing spondylitis), chronic infections (eg, bronchiectasis, pressure ulcers, subcutaneous drug use “skin popping,” osteomyelitis, and tuberculosis), inflammatory bowel disease, and rarely vasculitis are known associates of AA amyloidosis.18,37 A history of recurrent fevers, especially in the absence of infection, should prompt an evaluation for hereditary fever syndrome. Although lymphoproliferative disorders are usually associated with AL, 2 in particular are associated with AA. First is Castleman disease, also known as angiofollicular lymph node hyperplasia.38 Extensive involvement of the AA amyloid can occur, even with unicentric Castleman disease. Stabilization of symptoms has been reported after surgical resection. The other is IgM monoclonal gammopathy. This is usually associated with AL amyloidosis, but AA amyloidosis has also been reported.39 Hepato/splenomegaly in patients with Schnitzler syndrome (IgM monoclonal gammopathy, urticarial rash, arthralgia, bone pain, lymphadenopathy) should alert one toward the possibility of AA amyloidosis.40 AA amyloidosis has also been reported in patients with Waldenström macroglobulinemia without Schnitzler syndrome.41
Screening tests
Monoclonal protein testing of the serum and urine is readily available and should be performed on all suspected cases. AL amyloidosis is the most common and dangerous form of amyloidosis and definitely should be ruled out.42 In addition to serum and urine protein electrophoresis and immunofixation, serum free light chain test can increase the sensitivity (especially in monoclonal light chain only cases) by 10%-15%.43 Bone marrow biopsy should be performed if a monoclonal protein is identified or if the amyloid is AL or AH type. Erythrocyte sedimentation rate may be elevated in patients with a monoclonal protein and both erythrocyte sedimentation rate and C-reactive protein may be elevated in patients with a history of chronic inflammatory disease or infection. Serum amyloid A (SAA) level can also be directly measured. Although there is a good correlation between C-reactive protein and SAA, studies found that SAA is more sensitive at identifying patients at risk of development of AA and has better prognostic value.18,44 Unfortunately, the test is currently not approved in the United States.18
Biopsy
Although screening tests are helpful, amyloidosis is a histologic diagnosis and tissue biopsy is required.1 Other paraprotein related diseases can mimic amyloidosis. Nephrotic syndrome with monoclonal gammopathy occurs in monoclonal immunoglobulin deposition disease, immunotactoid glomerulonephritis, fibrillary glomerulonephritis, type I cryoglobulinemic glomerulonephritis, membranoproliferative glomerulonephritis, and others.45,46 Neuropathy and monoclonal gammopathy are seen in POEMS syndrome, IgM-associated neuropathy, monoclonal gammopathy of undetermined significance-associated neuropathies and anti-myelin–associated glycoprotein associated neuropathies.47,48 It is essential that amyloid deposition in tissue is demonstrated for the diagnosis.
In general, the highest sensitivity and specificity are achieved by obtaining tissue from the dysfunctional organ. Using this approach, studies have found heart to be 100% sensitive followed by liver at 97% and kidney at 94%. Although bleeding is a concern, the risk is no higher than patients without amyloidosis if standard screening procedure is followed.49 In situations where biopsy of the organ involved is not practical or unsafe, alternative sites may be biopsied. The easiest is abdominal fat. This can be done either by fine needle aspirate or biopsy. It has a sensitivity of ∼ 80%.16 Rectum is another commonly used site and has a sensitivity of 75%. It is usually performed via a flexible sigmoidoscopy. Bone marrow biopsy may also contain amyloid deposits, but sensitivity is only 56%. Amyloid can also be found in thyroid nodules, but incidence is unknown and there are no data on sensitivity. Given its safety, one should not hesitate to perform a fat aspirate if the index of suspicion is moderate or higher.
Histochemistry
On light microscopy with hematoxylin and eosin stain, amyloid appears as a light pink amorphous extracellular substance.50 It is negative on periodic acid-Schiff and methenamine silver stains, and blue or gray on trichrome stain. The gold standard for diagnosis of amyloid is the Congo red stain. All forms of amyloid exhibit affinity for Congo red and give off an apple-green birefringence under polarized light. It is one of the characteristics that distinguish amyloid from other fibrils. For best visualization of small amyloid deposits, the tissue sections for Congo red stain should be 6-10 μm in thickness (rather than the 2-4 μm thickness used for renal pathology and the 3-5 μm thickness for general pathology). Pay particular attentions to vessel walls where amyloid deposits can often be found in all tissue. The apple-green birefringence of amyloid should not be confused with the yellow-green birefringence of collagen.51 Although the birefringence is very specific to amyloid in animals, it occurs with cellulose. Thus it is important to avoid any cotton or paper fiber contamination on the slides. Thioflavin T, which binds to β-sheet rich structures, is another stain that can be used to diagnose amyloid. It is yellow but undergoes a red shift under fluorescence enhancement. Although it is sensitive, it is less specific than Congo red.
Electron microscopy
Electron microscopy is routinely used in analysis of kidney biopsy in United States and other countries. It is extremely helpful in the diagnosis because amyloid fibrils have unique electron microscopy characteristics. Amyloid fibrils are typically randomly arranged, nonbranching, and solid with a diameter between 7 and 12 nm.1 In comparison, fibrils of fibrillary and immunotactoid glomerulonephritis are larger at 7-26 nm and 9-45 nm, respectively.50 Fibrils of immunotactoid are similar to those of cryoglobulins and tend to be organized at least focally in parallel arrays. They also have a hollowed center resembling microtubules. The fibrils of these diseases typically stain for the entire immunoglobulin but rarely for a single light chain only. Most cases of immunotactoid glomerulonephritis and a minority of cases of fibrillary glomerulonephritis are associated with a monoclonal gammopathy. It is therefore not uncommon for them to be misdiagnosed as AH or ALH (immunoglobulin light and heavy chain) amyloidosis.52 Fibrils can also be found in hereditary nephropathies with fibronectin, and collagenofibrotic glomerulopathy, diabetic nephropathy (so called “diabetic fibrillosis”), lupus nephritis, and thrombotic microangiopathy.
Amyloid typing
Immunofluorescence and immunohistochemistry
Once amyloid has been identified, further characterization and typing must be performed. Direct immunofluorescence microscopy, which is routinely performed on native kidney biopsies, is one of the most common and convenient methods of amyloid typing and should be the first step. It is most useful in cases of amyloid composed of immunoglobulin components where antibodies are widely available. AL and/or AH amyloid can be typed by immunofluorescence using FITC-labeled antibodies to IgA, IgM, IgG, κ, and λ light chain. For AL amyloidosis, only one of the immunoglobulin light chains should stain positive in the amyloid deposits. For AH amyloidosis, the deposits will stain positive for a single subclass of immunoglobulin heavy chain without light chain, whereas a single light chain will also stain in ALH. Standard immunofluorescence is performed on fresh frozen tissue. In the absence of frozen tissue, immunofluorescence can be performed with pronase-digested paraffin-embedded tissue. Determining the immunoglobulin component involved in amyloidogenesis identifies the target of therapy. Recent studies found that reduction of the involved free light chain is more important than the intact immunoglobulin for patients with AL amyloidosis.53 An alternative method is immunohistochemistry using immunoperoxidase staining of formalin-fixed, paraffin-embedded tissue. This works well for AL, AH, AA, AFib, ATTR, AApoA1, and AB2m where antibodies are available commercially. Immunohistochemistry can also aid in the diagnosis of amyloidosis. Antibodies against serum amyloid P (SAP), which codeposits with amyloid, can be used to distinguish light chain deposits of AL amyloidosis from monoclonal immunoglobulin deposition disease. Immunofluorescence is superior to immunohistochemistry for the accurate typing of AL or AH amyloidosis because of the lower background staining in the former. Because frozen tissue is not usually available in nonrenal samples, immunohistochemistry, not immunofluorescence, is typically performed on these samples.
Despite the convenience and availability, there are some drawbacks to immunofluorescence and immunohistochemistry. One limitation is the availability of antibodies. Although antibodies are generally available for the most common forms of amyloid, availability is variable for the less common forms. Sensitivity and specificity of the antibodies are another problem. Because amyloid precursors often have genetic mutations and conformational changes, antibodies produced against the wild-type protein may be less reactive with the mutant form. This seems particularly problematic for AL, which has the greatest variability.5 In renal amyloidosis, the amyloid deposits can be negative for both κ and λ in 14% of cases using immunofluorescence.54 The same is true for immunohistochemistry. One study found 24 of the 76 samples suspected of AL could not be definitely diagnosed with immunohistochemistry.55 Thus, negative or equivocal staining for immunoglobulin light and/or heavy chains does not automatically rule out AL.55,56 Additional testing with a panel of antibodies directed toward other amyloidogenic proteins was required to make the diagnosis. Even with that, 5 patients could not be typed.55 On the other hand, better results were obtainable by the use of multiple antibodies. In a separate study with biopsies from patients with AL, AA, ATTR, AFib, and AApoA1, the sensitivity was increased to 92%-96% with a 100% specificity when multiple antibodies against κ and λ were used.57 In cases where the staining of light chains is negative or equivocal, the use of antibodies from different vendors and a panel of antibodies against other amyloids may be helpful. The biggest problem with these techniques, however, is the requirement to perform a separate test for each type of amyloid being considered. This becomes cumbersome once the common forms of amyloid have been ruled out and is impractical for the discovery of new type of amyloid. Finally, charge interaction of the amyloid and the reagent antibody or contamination of the amyloid deposits by serum proteins can all lead to nonspecific staining for immunoglobulins in immunohistochemistry or immunofluorescence. This nonspecific staining occurs more frequently in AA than AL amyloidosis.54–56
Source: American Society of Hematology
Read More